Acoplament molecular
Acoblament molecular (o docking) és una tècnica informàtica que intenta predir com dues molècules encaixen entre elles. En concret, calcula:
- Quina orientació adopta una molècula quan s’apropa a una altra.
- Quina forma exacta (conformació) és la més estable quan interactuen.
- I com de forta és aquesta unió.
Dit d’una manera més visual: imagina una clau i un pany. El docking intenta esbrinar quina és la millor manera d’introduir la clau al pany perquè encaixi bé i s’hi mantingui estable —sense forçar-la ni soldar-la.
En la pràctica, sovint es fa servir per estudiar com una molècula petita (per exemple, un possible fàrmac) s’uneix a una proteïna. Aquesta unió sol ser no covalent, és a dir, no implica enllaços químics permanents, sinó interaccions més febles (com ponts d’hidrogen, forces electrostàtiques o interaccions hidrofòbiques).
A més de predir la posició d’unió, el docking també estima:
- L’afinitat (com de forta és la unió).
- I, indirectament, la possible activitat biològica de la molècula.
Per això és una eina clau en la indústria farmacèutica: permet provar virtualment milers de molècules abans de sintetitzar-les al laboratori. Això accelera el disseny racional de fàrmacs, reduint temps, costos i intents a cegues.
En resum:
L’acoblament molecular és una simulació computacional que prediu com dues molècules s’encaixen i amb quina estabilitat, especialment útil per dissenyar nous medicaments.
Lligand
Un lligand és una substància que forma un complex amb una biomolècula per servir un propòsit biològic. L’etimologia prové del llatí ligare, que significa ‘unir’.
En la unió proteïna-lligand, el lligand és generalment una molècula que produeix un senyal en unir-se a un lloc específic d’una proteïna objectiu.
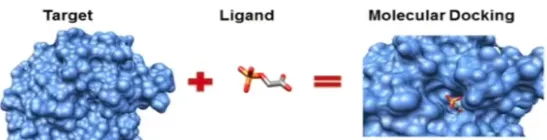
La unió típicament resulta en un canvi d’isomerisme conformacional (conformació) de la proteïna objectiu.
Quan alguna molècula —per exemple un fàrmac o un altre compost— s’uneix a una proteïna, aquesta proteïna canvia lleugerament la seva forma tridimensional. Quan canvia la forma pot activar-se, desactivar-se o fer una funció diferent.
Aplicacions del docking
1. Campanyes d’“in silico virtual screening” per combatre la COVID‑19
Durant la pandèmia, equips científics arreu del món van fer servir AutoDock Vina (un dels motors de docking més utilitzats) per buscar molècules que s’enganxessin al proteasa principal del SARS‑CoV‑2 — una estratègia essencial per proposar fàrmacs candidats abans d’entrar a laboratori. Aquestes simulacions es van fer amb pipelines automàtics (sovint controlats amb scripts i eines que s’integren amb Python) que carregaven molts compostos, els “docked” i puntuaven quins tenien més potencial.
- ❗ Aquesta cerca va revelar molècules anticovid amb bona afinidad de unió al SARS‑CoV‑2, i això després es va correlacionar amb estudis biològics reals en cultius cel·lulars.
- 🧪 Aquests resultats van inspirar camps de recerca i disseny de fàrmacs que avui encara s’expliquen en articles i tesis.
Font: Estudis clínics i acadèmics publicats analitzant docking contra les proteïnes clau de SARS‑CoV‑2.
2. FightAIDS@Home — recerca distribuïda per combatre l’HIV
Aquest projecte és emocionant perquè involucra milers de voluntaris de tot el món:
FightAIDS@Home és una iniciativa que utilitza computació distribuïda i software de docking (AutoDock VINA) per analitzar milions de possibles molècules que podrien inhibir la proteasa del VIH, un objectiu clau per aturar la replicació del virus.
Després de cribratges computacionals extensius, es van identificar nous compostos que actuen sobre llocs d’unió no explotats del virus, obrir potencials fàrmacs per combatre resistències del VIH.
Aquest projecte no només va generar coneixement nou, sinó que ha inspirat biomèdica real i papers científics. FightAIDS Home - Wikipedia
Passos a seguir.
El pipeline (seqüència de passos i algorismes) per a realitzar el docking de forma automatitzada ha d’estar ben definit en passos, per a poder identificar i resoldre qualsevol imprevist.
1. Definir el problema
- Proteïna, binding site (lloc d’unió), objectiu.
Sense això, tot el pipeline és soroll.
2. Preparar el receptor
- Neteja (aigües, ligands)
- Afegir hidrogens i càrregues
Eines Python recomanades:
- Scripts Python per tractar fitxers PDB (molt còmode en Python)
- Biopython (manipulació d’estructures, avançat)
Font fitxers PDB en 3D: PDBank.
3. Preparar el ligand
- 3D, protonació, tautòmers, conformers
Eines Python:
- Scripts Python per tractar fitxers SDF (molt còmode en Python)
- RDKit per convertir SMILES en 3D i optimitzar estructures (només si no està preparada)
Font fitxers SDF/MOL en 3D: Pubchem.
4. Definir l’espai de cerca
- Centre i mida del binding site. El binding site és una caixa 3D on veiem el tros de mol·lècula on ens interessa acoplar el lligand.
- Cal ser molt específics per obtenir bons resultats i un rendiment viable.
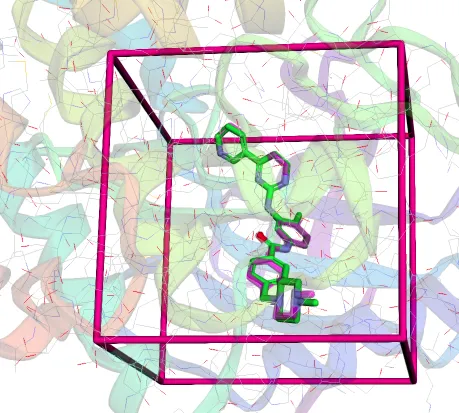
Sovint:
- scripts Python simples
- visualització amb PyMOL
5. Preparació per docking
- Convertir a format compatible.
Eines Python:
- Meeko per generar PDBQT directament.
- OpenBabel també s’usa molt per aquest propòsit però és menys automatitzable.
6. Docking (sampling i scoring)
- Explorar poses
- Calcular energies
Eines Python:
- AutoDock Vina amb API Python.
Aquesta part requereix instal·lació de software al Sistema Operatiu, la compatibilitat és més alta en Linux.
7. Ranking i resultats
- Selecció de millors poses (es proven moltes posicions x,y,z i es trien les millors)
Eines Python:
- parsing amb scripts propis
- pandas/polars per analitzar energies
8. Anàlisi i validació
- Interaccions, plausibilitat
Eines Python:
- PyMOL per a visualitzar com ha quedat.
- MDAnalysis
Resum compacte
Receptor → Fitxers PDB. Es poden editar amb Python o Prody.Ligand → Fitxers SDF o MOL. Es poden editar amb RDKit si cal.Prep → MeekoDocking → AutoDock VinaAnàlisi → PyMOLAmb Python, el docking deixa de ser una seqüència manual i es converteix en:
un pipeline automatitzable, reproduïble i escalable
I això canvia completament el joc.
Python Tools
Python offers a range of libraries and tools that facilitate molecular docking simulations and analysis. Some popular Python libraries used in molecular docking are:
-
PyAutodock: PyAutodock is a Python wrapper for the popular docking software AutoDock Vina. It allows you to perform molecular docking studies using AutoDock Vina from within Python and enables programmatic control of the docking process.
-
PyRx (Python Prescription): PyRx is a virtual screening tool that utilizes Autodock Vina under the hood. It provides a user-friendly graphical interface but also offers a Python API for more advanced users who want to automate tasks or perform custom analyses.
-
Open Babel: Open Babel is a chemical toolbox designed to speak the many languages of chemical data. It can be used to convert molecular file formats and manipulate molecular structures, which is often useful in preparing input files for docking simulations.
-
RDKit: RDKit is another versatile cheminformatics library that can handle molecular data, including 2D and 3D molecule rendering, chemical transformations, and property calculations, which can be beneficial in preparing ligands and protein structures for docking studies.
These libraries provide functionalities to set up docking simulations, analyze docking results, visualize interactions, and perform further analysis. Additionally, with Python’s extensive scientific ecosystem, you can combine these libraries with others for data manipulation, visualization, and statistical analysis, making it a popular choice for researchers and scientists in the field of molecular docking.