Alineament de seqüències
L'alineament de seqüències és un mètode computacional que permet inferir informació biològica només a partir de la informació que tenen les seqüències.
Introducció
La forma més fiable de determinar l’estructura o funció d’una molècula biològica és mitjançant l’experimentació directa. No obstant això, és molt més fàcil obtenir la seqüència d’ADN del gen corresponent a un ARN o una proteïna, que determinar experimentalment la seva funció o estructura.
Per tant, part del repte és simplement organitzar, classificar i analitzar la immensa riquesa de dades de seqüències, i desenvolupar mètodes computacionals que puguin inferir informació biològica només a partir de la seqüència.
Seqüències
Per entendre la natura no has de pensar en ella com un inventor que dissenya els organismes, sinó com una “entitat” que es dedica a arreglar i experimentar amb organismes i els seus components.
Les noves seqüències s’adapten a partir de seqüències preexistents en lloc d’inventar-se de zero, la qual cosa ens permet analitzar les seqüències de forma computacional per reconèixer una similitud significativa entre una seqüència nova i una seqüència sobre la qual ja se sap alguna cosa.
Quan fem això podem transferir informació sobre l’estructura i/o la funció a la nova seqüència. Diem que les dues seqüències relacionades són homòlogues i que estem transferint informació per homologia.
Dues seqüències són homòlogues si es deriven d’una seqüència comuna, és a dir, tenen un ancestre comú.
L’homologia no és quantificable, les seqüències són o no són homòlogues.
Per tant, una de les preguntes més bàsiques sobre un gen o proteïna és si està relacionat amb algun altre gen o proteïna:
-
La relació de dues proteïnes a nivell de seqüència suggereix que són homòlogues.
-
La relació també suggereix que poden tenir funcions comunes.
Mitjançant l’anàlisi de moltes seqüències d’ADN i proteïnes, és possible identificar dominis o motius que es comparteixen entre un grup de molècules.
Aquestes anàlisis de la relació de proteïnes i gens s’aconsegueixen alineant seqüències.
La majoria dels problemes en l’anàlisi de seqüències computacionals són essencialment estadístics. Les forces evolutives estocàstiques actuen sobre els genomes. Discernir similituds significatives entre seqüències antigament divergents enmig d’un caos de mutació aleatòria, selecció natural i deriva genètica presenta seriosos problemes de senyal al soroll. Molts dels mètodes d’anàlisi més potents disponibles utilitzen la teoria de la probabilitat.
Alineament de proteïnes
A l’hora de comparar proteïnes, podem comparar la seqüència d’aminoàcids que la componen, o comparar els gens que codifiquen la seqüència d’aminoàcids.
Les seqüències d’aminoàcids són més informatives perquè:
-
Molts canvis en una seqüència d’ADN (especialment en la tercera posició d’un codó) no canvien l’aminoàcid que s’especifica.
-
Molts aminoàcids comparteixen propietats biofísiques relacionades (per exemple, la lisina i l’arginina són ambdós aminoàcids bàsics).
El resultat és que les comparacions de seqüències de proteïnes poden identificar seqüències homòlogues mentre que les comparacions de seqüències d’ADN corresponents no poden.
Per tant, quan s’analitza una seqüència de codificació de nucleòtids sovint és preferible estudiar la seva proteïna traduïda.
A Blast veurem que podem moure’ns fàcilment entre els mons de l’ADN i les proteïnes.
Per exemple, l’eina TBLASTN del lloc web NCBI BLAST permet cercar proteïnes relacionades derivades d’una base de dades d’ADN amb una seqüència de proteïnes.
Aquesta opció de consulta s’aconsegueix traduint cada seqüència d’ADN a les sis proteïnes que potencialment codifica.
No obstant això, tot el que hem explicat fins ara, en molts casos és adequat comparar seqüències d’ADN.
Aquesta comparació pot ser important per confirmar la identitat d’una seqüència d’ADN en una cerca de bases de dades, per cercar polimorfismes, per analitzar la identitat d’un fragment d’ADNc clonat o per comparar regions reguladores, per posar alguns exemples.
Homología
Dos secuencias son homólogas si comparten un ascendente evolutivo común.
No existen grados de homología; las secuencias son homólogas o no.
Las proteínas homólogas casi siempre comparten una estructura tridimensional significativamente relacionada. Un ejemplo de homología es el de la mioglobina humana (NP_005359.1) y la beta globina (NP_000509.1), dos proteínas que están relacionadas lejanamente, pero significativamente.
Se cree que la mioglobina y las cadenas de hemoglobina (alfa, beta y otros) divergieron hace unos 450 millones de años, cerca del momento en que los linajes de peces humanos y cartílagos divergieron.
La mioglobina y la beta globina tienen estructuras muy parecidas, tal y como determina la cristalografía de rayos X tal y como puedes ver en esta imagen:
| Human myoglobin (3RGK) | Human beta globin (subunit of 2H35) |
|---|---|
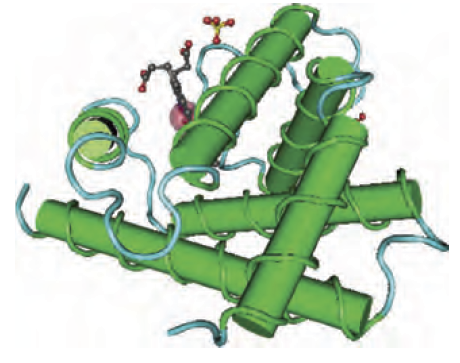 | 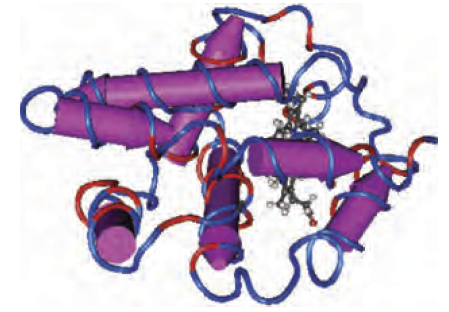 |
Estas proteínas son homólogas (descienden de un antepasado común) y comparten estructuras tridimensionales muy similares.
Sin embargo, el alineamiento por parejas de las secuencias de aminoácidos de estas proteínas revela que las proteínas comparten una identidad de aminoácidos muy limitada.
Identidad
Cuando dos secuencias son homólogas, sus secuencias de aminoácidos o de nucleótidos suelen compartir una identidad significativa.
Si bien la homología es una inferencia cualitativa (las secuencias son o no homólogas), la identidad y la semejanza son cantidades que describen la relación de las secuencias.
En particular, dos moléculas pueden ser homólogas sin compartir una identidad estadísticamente significativa de aminoácidos (o nucleótidos).
En la familia de la globina, todos los miembros son homólogos, pero algunos tienen secuencias que tanto han divergido que no comparten ninguna identidad de secuencia reconocible
Por ejemplo:
- La beta globina humana y la neuroglobina humana sólo comparten un 22% de identidad de aminoácidos.
- Las cadenas de globina individuales comparten la misma forma general que la mioglobina, aunque las proteínas de mioglobina y alfa globina sólo comparten un 26% de identidad de aminoácidos.
Por lo general, las estructuras tridimensionales divergen mucho más lentamente que la identidad de la secuencia de aminoácidos entre dos proteínas.
¡Y es la estructura la que define la función de la proteína!
Reconocer este tipo de homología es un problema bioinformático especialmente complejo.
Ortología
Las proteínas que son homólogas pueden ser ortologues o paralogas.
Los ortólogos son secuencias homólogas en distintas especies que tienen un gen antepasado común y que han divergido por especiación.
La historia del gen refleje la historia de la especie, por lo que los genes se llaman ortólogos (orto = exacto). Se supone que los ortólogos tienen funciones biológicas similares, pudiendo deducir la función de una secuencia a partir de otra secuencia homóloga.
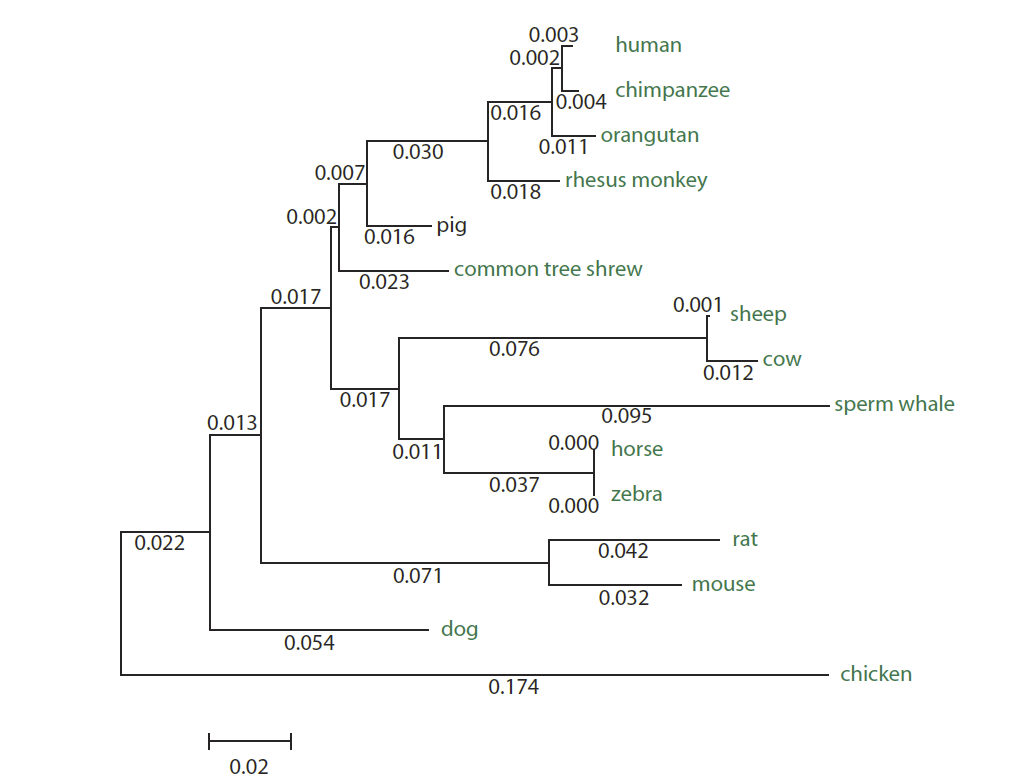
Por ejemplo, los humanos y roedores divergieron hace unos 90 millones de años (MYA), momento en que un solo gen de la mioglobina ancestral divergió por especiación, y en ambos casos la función del gen es transportar oxígeno a las células musculares
A continuación tienes un dibujo con el árbol de los ortólogos de la mioglobina:
Paralogía
Los parálogos son secuencias homólogas surgidas por un mecanismo como la duplicación de genes.
En este caso la homología es el resultado de la duplicación génica por lo que ambas copias han bajado una al lado de la otra durante la historia de un organismo.
Los genes se llaman paralógicos (para = en paralelo) porque evolucionan dentro de la misma especie.
Por ejemplo, la globina alfa 1 humana (NP_000549.1) es paraloga en la globina alfa 2 (NP_000508.1). De hecho, estas dos proteínas comparten una identidad de aminoácidos del 100%.
La globina alfa 1 y la globina beta humanas también son paralogos, como todas las proteínas humanas que se muestran a continuación y que son miembros de la familia globina:
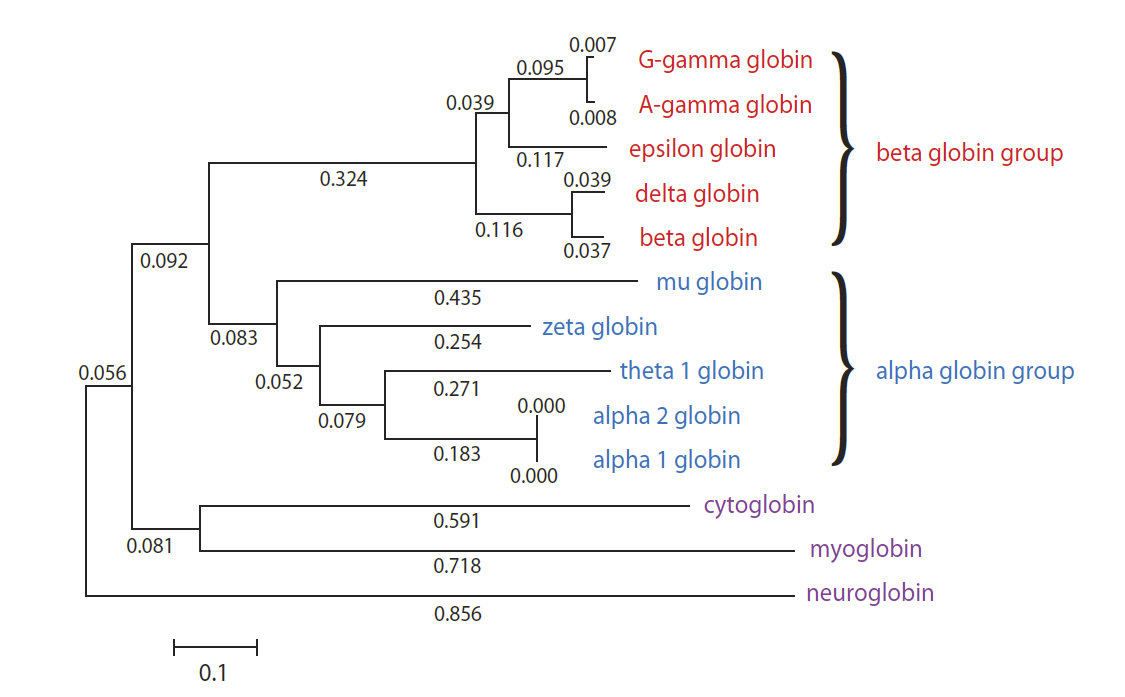
Todas las globinas tienen propiedades distintas, incluida la distribución regional en el cuerpo, el tiempo de desarrollo de la expresión génica y la abundancia. Pero aunque todas tienen funciones distintas, estas funciones están relacionadas como proteínas portadoras de oxígeno.
Para saber un poco más sobre estos árboles y la evolución puede consultar este video:
Similitud
Ver “/python/dynamic-programming”
A primera vista, decidir que dos secuencias biológicas son similares no es diferente decidir que dos cadenas de texto son similares.
Los algoritmos de semejanza de texto se utilizan para buscar la palabra más parecida en un diccionario cuando una palabra no se encuentra en un diccionario porque está mal escrita. Es lo que conoces como corrector ortográfico básico.
Por tanto, no debe extrañar que los algoritmos de análisis de secuencias compartan mucho parecido con los algoritmos de semejanza de texto.
La distancia de Levenshtein (o distancia de edición) es un algoritmo de 1965 diseñado para comparar dos palabras, que se aplicó posteriormente a secuencias biológicas.
Este algoritmo se basa en que, para transformar una palabra en otra, sólo son necesarias tres operaciones básicas: insertar, suprimir o sustituir un carácter.
Cada vez que se aplica una de estas operaciones, la distancia se incrementa 1 punto, y cuantos más puntos mayor es la distancia de edición de las dos palabras y menos similares son.
Matriz de distancias
La distancia de edición es un algoritmo lineal que utiliza una matriz (un array de dos dimensiones) para guardar los resultados parciales que permiten ir construyendo el resultado final.
Dadas dos secuencias de ácidos nucleicos xs y ys, la matriz de distancias contiene las distancias entre todos los prefijos de la secuencia xs y todos los prefijos de la secuencia ys.
Para empezar, debes llenar la matriz con el resultado del subproblema básico a partir del cual iremos construyendo resultados parciales hasta llegar al resultado final.
Por ejemplo, si quieres comparar las secuencias CGA y AGAT, debes crear una matriz de 4 x 5 con estos valores iniciales (el símbolo - indica un string vacío "")
Como puedes ver en este ejemplo, es evidente que la distancia de edición entre:
- “” i “A” es 1, porque para transformar “” en “A” debemos insertar una A.
- “” i “EN” es 2, porque para transformar “” en “EN” debemos insertar una A y después una G.
A continuación tienes el algoritmo inicial implementado en Python:
=
=
=
=
=
Si ejecutamos el script podemos ver la tabla resultante:
Estàs llegint una vista prèvia.
Inicia sessió amb Google per llegir la pàgina completa. Segueix l'itinerari d'aprenentatge — cada pàgina es desbloqueja quan has llegit les que la precedeixen. L'alumnat i el professorat llegeixen les pàgines del seu curs sense límit.
Inicia sessió